Chamada Lilypad, a sua construção foi inspirada num nenúfar gigante descoberto na Amazónia por Thaddeaus Haenke, no início do século XIX. O botânico alemão baptizou-o de Vitória régia, em homenagem à rainha Vitória de Inglaterra...
Pânico ecológico - Humanidade precisará de dois planetas em 2030.
Com o actual ritmo de consumo dos recursos naturais do nosso planeta, segundo o relatório Planeta Vivo de há dois anos - responsabilidade da organização WWF, Sociedade Zoológica de Londres e da Global Footprint Network - precisaríamos de um segundo planeta por volta do ano 2050...
A China vista dos Céus.
A China não cessa de nos surpreender; a fotografia aérea também, ao revelar-nos formas, cores e texturas improváveis que nos dão uma outra noção do espaço. Este conjunto de fotografias aéreas da China põe em evidência o contraste entre a dimensão humana e a vastidão do imenso território chinês...
O Natal, o Papai Noel e a Coca-Cola.
A lenda do Papai Noel (Pai Natal em Portugal) é inspirada no arcebispo São Nicolau Taumaturgo, que viveu na Turquia no século IV. Ele tinha o costume de ajudar os necessitados depositando um pequeno saco com moedas de ouro, entrando nas casas pela lareira...
Publicidade - Os direitos dos animais.
Criatividade e consciencialização são palavras de ordem na nova campanha publicitária realizada pela agência WCRS, que assina Born Free “Keep wildlife in the Wild”. Qualquer um de nós tem consciência da quantidade de pessoas, que por falta de recursos ou alternativas, vivem nas ruas. A última campanha da Born Free, pega nesta ideia e coloca animais selvagens, sem lar, em cenários urbanos...
Os portadores de fenilcetonúria não conseguem metabolizar a fenilalanina, portanto o excesso dessas moléculas no sangue se transforma num ácido fenilpirúvico, que exerce ação tóxica em vários órgãos, especialmente no cérebro.
Fenilcetonúria (PKU, na sigla em inglês) é uma doença decorrente de um erro inato do metabolismo de aminoácidos, que ocorre por herança autossômica recessiva. Isso significa que a criança precisa receber um gene alterado da mãe e outro do pai para desenvolver o distúrbio.
A mutação nesse gene provoca um defeito na codificação de uma enzima, a fenilalanina hidroxilase (PAH), necessária para transformar o aminoácido essencial fenilalanina – que o organismo não produz, mas retira dos alimentos proteicos – em tirosina, outro aminoácido importante para a estrutura das proteínas.
Como os portadores de fenilcetonúria não conseguem metabolizar a fenilalanina, o excesso dessas moléculas no sangue se transforma num ácido fenilpirúvico, que exerce ação tóxica em vários órgãos, especialmente no cérebro.
SINTOMAS
Recém-nascidos portadores do distúrbio são assintomáticos. Sem tratamento, porém, logo nos primeiros meses de vida, começam a surgir os seguintes sinais da doença, que podem variar de intensidade conforme o caso:
Odor no suor e na urina parecido com o do bolor ou da urina de rato;
Pele, cabelos e olhos claros, porque a fenilalanina não se converte em tirosina, aminoácido envolvido na síntese de melanina, o pigmento que dá cor à pele.
CLASSIFICAÇÃO
De acordo com o erro metabólico envolvido, que se reflete sobre a atividade maior ou menor da enzima fenilalanina hidroxilase (PAH), e os níveis plasmáticos de fenilalanina (FAL), a doença admite a seguinte classificação:
Fenilcetonúria clássica – enzima quase ausente e níveis plasmáticos de FAL elevados – é a forma mais grave da doença; sem tratamento, o portador desenvolve lesões cerebrais irreversíveis;
Fenilcetonúria leve – índices moderados de um e outro componente – os pacientes dependem do tratamento para controlar a evolução da doença;
Hiperfenilalninemia transitória ou permanente – atividade superior de PAH e inferior de FAL – quadro benigno sem manifestações clínicas.
As leucodistrofias são doenças progressivas, geneticamente determinadas, que afetam o sistema nervoso em decorrência de alterações na bainha de mielina (estrutura que reveste as células nervosas).
Muitos são os genes que atuam nos diferentes passos do processo de mielinização dos neurônios. Em virtude da heterogeneidade genética, o grupo das leucodistrofias comporta vários padrões de herança, como a herança autossômica recessiva e dominante, além da herança ligada ao cromossomo X. Cada um destes padrões de herança corresponde a riscos de recorrência da doença muito diferentes, o que reforça a importância do aconselhamento genético para o paciente e seus familiares.
Na maioria dos casos, a leucodistrofia é herdada, mas há relatos de mutação de novo. Nesta situação, o risco de recorrência na irmandade de um afetado é desprezível.
Quadro Clínico:
As leucodistrofias constituem um grupo heterogêneo de doenças, com grande variabilidade clínica. Esta variabilidade reflete diferenças na etiologia genética, que levam ao comprometimento de regiões do sistema nervoso específicas para cada leucodistrofia.
Em geral, os sinais clínicos se manifestam após um período de vida livre de qualquer sintoma. Nos estágios iniciais, os sinais clínicos são pouco reconhecidos. As manifestações ocorrem de maneira gradual, sendo paulatinamente notadas como: mudança no tônus corporal e nos movimentos, alterações de marcha, fala, visão, audição, comportamento, memória e processos mentais.
O diagnóstico específico depende de exames de imagem, como a ressonância magnética e a tomografia computadorizada, e da realização de análises bioquímicas. Entre estas, podem ser indicadas, por exemplo, a dosagem de ácidos graxos de cadeia muito longa (cujos níveis encontram-se elevados na leucodistrofia chamada de adrenoleucodistrofia ligada ao cromossomo X) e a determinação da atividade de algumas enzimas lisossomais (como a arilsulfatase A, que apresenta níveis anormais na leucodistrofia metacromática, e a galactocerebrosidase, alterada na Doença de Krabbe).
Exemplos de leucodistrofias:
Adrenoleucodistrofia
Adrenoleucodistrofia ligada ao X
Doença de Alexander
Doença de Canavan
Doença de Krabbe
Doença de Pelizaeus Merzbacher (paraplegia espástica ligada ao X)
Doença de Refsum
Leucodistrofia metacromática
Síndrome 18q com deficiência de proteína básica mielina
A Doença de Huntington (DH) é uma afecção heredodegenerativa (isto é, herdada geneticamente e progressiva) do sistema nervoso central, cujos sintomas são causados pela perda marcante de células em uma parte do cérebro denominada gânglios da base. Esse dano no cérebro afeta as capacidades:
A DH atinge homens e mulheres de todas as raças e grupos étnicos e, de forma geral, os primeiros sintomas aparecem lenta e gradualmente entre os 30 e 50 anos, mas pode atingir também crianças (veja DH Juvenil) e idosos.
Filhos que tenham um dos pais afetado pela DH têm 50% de chances de herdar o gene alterado e desenvolverão a doença em algum momento de sua vida.
Não existe ainda cura ou tratamento eficaz para a DH, mas, na maior parte dos casos, as pessoas podem manter sua independência por vários anos após o aparecimento dos primeiros sintomas da doença. Um médico bem informado pode prescrever um tratamento para minimizar o impacto dos sintomas motores e mentais, embora estes sejam progressivos.
Outros profissionais de saúde, tais como fisioterapeuta, terapeuta ocupacional, fonoaudiólogo, nutricionista, psicólogo e assistente social, podem ajudar muito na maximização das habilidades e prolongamento da independência do paciente de Huntington, proporcionando melhor qualidade de vida tanto para o paciente quanto para a família.
Desde que, em 1872, Dr. George Huntington publicou o primeiro artigo sobre a DH, a que ele chamou de “Coreia Hereditária”, vários avanços aconteceram. A partir de 1970 os estudos sobre a DH começaram a se intensificar: em 1983 foi descoberto o defeito genético responsável pela doença e em 1993 foi isolado o gene, tornando-se possível os testes genéticos e as pesquisas em animais, o que desencadeou pesquisas importantes.
Vários estudos colaborativos existem desde então e a partir de 2008 a CHDI Foundation (http://www.chdifoundation.org/), mantida por famílias com DH, que tem como missão “Descobrir rapidamente medicamentos que retardem o início ou a progressão da doença de Huntington”, faz grandes investimentos em parceria com laboratórios e centros de pesquisas. Este é um grande diferencial em relação a outras doenças e grande alento e esperança para as famílias com DH em todo o mundo.
Perguntas Frequentes sobre a DH
1. O que é a Doença de Huntington?
A Doença de Huntington (DH), também conhecida como Coreia de Huntington, é uma doença hereditária rara, neurodegenerativa, que afeta o sistema nervoso central, causando alterações dos movimentos, do comportamento e da capacidade cognitiva.
2. Por que se chama Doença de Huntington?
A DH recebeu o nome de George Huntington, um médico americano que descreveu a doença, em 1872. A sua descrição baseou-se em observações de famílias afetadas pela DH no povoado de East Hampton, Long Island, Nova Iorque (Estados Unidos da América), onde Huntington morava e trabalhava como médico. Ele foi a primeira pessoa a identificar o padrão hereditário da DH.
No passado a DH já foi conhecida, popularmente, por “Dança de São Vito e Dança de São Guido”.
3. O que causa a Doença de Huntington?
A DH é causada por uma mutação no gene que codifica uma proteína chamada huntingtina (Htt). Esta mutação produz uma forma alterada da proteína Htt, que causa a morte das células nervosas (neurônios) em determinadas regiões do cérebro.
4. De que modo a mutação causa a morte das células nervosas?
O mecanismo exato da doença ainda não foi esclarecido. Dois caminhos foram propostos: No primeiro, a proteína não pode exercer mais a sua função normal (perda de função). No segundo caso, a proteína mutada pode ser tóxica para as células nervosas (ganho de função).
5. O que acontece com o cérebro durante a DH?
Certas funções do cérebro, como a habilidade de se mover, pensar e falar, deterioram-se gradualmente à medida que células nervosas chaves são danificadas e morrem. A parte do cérebro mais afetada pela DH é o corpo estriado, que é uma estrutura dos gânglios da base localizada na região central do cérebro. O corpo estriado é composto de três regiões, denominadas núcleo caudado, putamen e núcleo accumbens.
O corpo estriado é basicamente responsável pelo planejamento e controle dos movimentos, mas também está envolvido em outros processos cognitivos (intelectuais). À medida que a doença progride, ocorre uma destruição do córtex cerebral (massa cinzenta correspondente à camada mais externa do cérebro) que contribui para a deterioração da capacidade cognitiva.
6. Quando surgem os sintomas da DH?
A maioria das pessoas afetadas desenvolve a doença durante a meia-idade, isto é, entre 35 e 55 anos. Aproximadamente 10% das pessoas desenvolvem sintomas antes dos 20 anos (DH juvenil) e outros 10% depois dos 55 anos. Mais raramente, os sintomas podem aparecer antes dos 10 anos de idade (DH infantil).
7. Quanto tempo dura a DH?
A DH é uma doença fatal que se desenvolve gradualmente. A duração média da doença é de 15 a 20 anos, mas isto varia de pessoa para pessoa.
8. A DH causa a morte?
A maioria das pessoas com DH não morre como resultado imediato da doença, mas sim de complicações causadas pelo estado de fragilidade do corpo, particularmente por asfixia em consequência de o doente se engasgar, por infecções (por exemplo pneumonia) e por parada cardíaca.
9. Como posso saber se tenho a DH?
Se você suspeita que tem a DH, você deve consultar um especialista em DH (normalmente um neurologista) para que seja feito o diagnóstico.
10. Qual é a incidência da DH?
A DH é uma doença rara que afeta até 1 em cada 10.000 pessoas na maioria dos países europeus. Na Alemanha por exemplo, aproximadamente 10.000 pessoas têm DH e outras 50.000 pertencem ao grupo de risco porque um dos seus pais tem (ou tiveram) DH. Homens e mulheres podem igualmente herdar o gene e desenvolver a doença.
11. A DH ocorre com a mesma frequência em países diferentes?
A DH pode afetar pessoas de todos os grupos étnicos, embora seja mais comum entre descendentes europeus. A incidência da DH em países predominantemente de origem europeia (como por exemplo EUA, Canadá e Austrália) é similar à da Europa.
Nos EUA por exemplo, aproximadamente 30.000 pessoas tem DH e outras 150.000 são consideradas em risco. A DH é menos comum em países asiáticos e africanos, onde a sua frequência foi estimada em 1 em cada 1.000.000 de pessoas. Contudo, estudos detalhados ainda não foram conduzidos nestes países, com exceção do Japão, onde se sabe que existem menos casos de DH do que na Europa.
Não existem estatísticas oficiais no Brasil, mas estima-se que sejam de 13.000 a 19.000 portadores do gene e de 65.000 a 95.000 pessoas em risco (seus descendentes).
eja por crença, solidariedade com os animais ou modismo, não ingerir carne ou alimentos de origem animal (dieta vegana) é uma tendência que vem ganhando cada vez mais força nos últimos anos.
No Reino Unido, por exemplo, o número de pessoas que se tornam veganas aumentou 350% na última década.
E isso vem alarmando alguns especialistas, sobretudo quando se adota essa dieta por puro modismo.
"Quando as pessoas se tornam veganas devido a alguma religião, tendem a saber como fazê-lo corretamente. Em outras palavras: sabem como compensar as deficiências nutricionais decorrentes da não ingestão de carne e produtos lácteos", diz Catherine Collins, da Associação Dietética Britânica.
"Mas não temos observado isso com pessoas que se tornam veganas para ficarem parecidas com outras pessoas que veem na internet", acrescenta.
Celebridades como a atriz americana Gwyneth Paltrow tornaram esse tipo de dieta popular.
"Essas pessoas veem fotos de Gwyneth Paltrow e querem fazer a mesma coisa. Querem estar na moda, mas estão colocando em risco sua própria saúde a longo prazo".
A dieta vegana exclui qualquer alimentos de origem animal, como carne, peixe, leite, queijo, ovos e mel.
Há veganos ainda mais ortodoxos que não cozinham verduras. Eles seguem uma dieta conhecida como crudiveganismo.
Muitos defensores, no entanto, dizem que a dieta traz uma série de benefícios para a saúde. No entanto, especialistas em nutrição alertam para a falta de proteínas, vitaminas e nutrientes importantes.
Em entrevista à BBC Mundo, o serviço em espanhol da BBC, Jesús Román, presidente da Sociedade Espanhola de Nutrição e Ciências da Alimentação, disse que a dieta vegana "é complicada e deve ser adotada com conhecimentos suficientes".
"Quando um grupo de alimentos é eliminado, neste caso de forma muito ampla, uma vez que são descartados todos os alimentos de origem animal, há uma série de nutrientes que são muito difíceis de se obter comendo apenas verduras", explica.
Vitamina B-12
A vitamina B-12 é essencial para o desenvolvimento de glóbulos vermelhos saudáveis e pode ajudar a prevenir a anemia.
O problema é que ela só é encontrada em produtos de origem animal - e em alguns poucos tipos de algas.
"Para que possa sobreviver por muito tempo, dado que a carência da vitamina B-12 demora para se manifestar, um vegano deve ingerir suplementos dessa vitamina".
Proteínas
A proteína ajuda a construir e manter saudáveis os músculos, os órgãos, a pele e os ossos.
Se você ingere exclusivamente verduras e legumes, especialistas recomendam incluir na dieta determinados produtos como frutas secas, sementes, produtos à base de soja, legumes, lentinhas e grãos, de forma a obter suficiente proteína.
"A proteína é um elemento chave na dieta de uma pessoa. Para se obter proteína de qualidade em uma dieta vegana, é preciso misturar diferentes alimentos - cereais com legumes por exemplo", explicou Román.
Direito de imagemTHINKSTOCKImage captionFerro é vital para a energia e para o correto funcionamento dos glóbulos vermelhos
Ferro, cálcio e ácidos graxos Ômega-3
O ferro é vital para a energia e para o correto funcionamento dos glóbulos vermelhos.
Mas, segundo o especialista, é difícil ingerir ferro suficiente em uma dieta vegetariana, já que ele é absorvido mais facilmente a partir de produtos de origem animal do que vegetal.
Assim, um vegetariano deve comer grandes porções de folhas verdes escuras, cereais integrais, feijão ou ervilha, lentilha, cereais enriquecidos e frutas secas.
Para aumentar a absorção de ferro, recomenda-se também comer alimentos ricos em vitamina C, como repolho, tomate, brócolis, morangos e limões.
Por outro lado, o cálcio é importante para manter os ossos e dentes fortes. Dessa forma, especialistas recomendam que, na eventualidade de cortarem os laticínios da dieta, os adeptos da dieta vegana usem substitutos como o leito de soja ou suco de frutas, cereais ou mesmo tofu com adição de cálcio.
Folhas verde-escuras, como brócolis e couve, também contêm cálcio, mas sozinhas não são suficientes para suprir a necessidade de cálcio do organismo.
Já os ácidos graxos ômega-3 ajudam a promover o tecido saudável, o desenvolvimento do olho e do cérebro, e a saúde cardiovascular.
Sem ovos ou peixes na dieta, a recomendação é buscar produtos enriquecidos com ácidos graxos ômega-3. Mas, segundo especialistas, talvez seja preciso ingerir um suplemento nutricional para suprir essa carência.
Román destaca ainda que o primeiro passo antes de adotar uma dieta vegana é estar saudável.
Ele também recomenda que os adeptos dessa dieta se submetam a exames de sangue regularmente para determinar se há carência de algum nutriente em especial.
*Após a publicação desta reportagem, a Sociedade Vegetariana Brasileira questionou algumas conclusões dos especialistas citados, argumentando que "quando a dieta vegetariana é seguida no longo prazo, reduzimos as taxas de mortalidade de doenças cardiovasculares, diabetes, câncer e obesidade, que são fatores que colocam a qualidade e expectativa de vida em risco. Além disso, sendo a dieta vegetariana adotada por pessoas no mundo há milênios, como na Índia, não é correto dizer que é uma dieta de moda."
A Organização diz ainda que "consumo de linhaça e chia fornece ômega-3 em abundância na dieta vegetariana" e que "praticamente todos os estudos que comparam a ingestão de ferro entre vegetarianos e não vegetarianos demonstram maior ingestão do mineral pelos vegetarianos. Além disso, a prevalência de anemia nas duas dietas não é diferente. A fração de ferro obtida pela carne não vai corresponder nem a 20% da recomendação diária de ingestão".
As pessoas que adotam uma dieta vegana, que exclui a carne ou qualquer alimento de origem animal, devem, entre outros cuidados, se certificar de que estão consumindo quantidade suficiente de um nutriente fundamental, mas pouco conhecido, para o cérebro.
A colina, que ajuda na comunicação entre células nervosas, é encontrada em maior concentração em carnes e laticínios.
E quem não ingere esse tipo de alimento corre o risco de não obter colina suficiente, alerta a nutricionista Emma Derbyshire em artigo publicado na revista científica BMJ Nutrition, Prevention & Health.
O nutriente, que também está associado à função hepática, está presente principalmente no ovo, no leite e na carne.
Mas existem alternativas. A colina também pode ser obtida pela ingestão de soja torrada, vegetais crucíferos - como brócolis e couve-de-bruxelas -, feijão cozido, cogumelos, quinoa e amendoim.
Derbyshire, especialista independente em nutrição e ciências biomédicas, escreveu na publicação científica que o Reino Unido estava ficando para trás de outros países por não recomendar ou monitorar os níveis de ingestão de colina.
As autoridades de saúde dos EUA, por exemplo, definem como níveis de "ingestão adequada" 425 mg/dia para mulheres e 550 mg/dia para homens.
Adotar o estilo vegano é uma escolha pessoal, mas grande parte dos seguidores atribui a opção a questões éticas e à preocupação com o meio ambiente. E há quem cite questões de saúde.
A colina, que faz parte das vitaminas do complexo B, é considerada essencial para a síntese do neurotransmissor acetilcolina, que desempenha um papel importante nas funções cognitivas, como processos de aprendizagem e formação de memórias.
E é particularmente importante para as mulheres durante a gravidez e a amamentação, uma vez que é transportada ativamente para o feto no útero, com suprimentos maternos relacionados à cognição, ou transferida para o bebê por meio do leite materno.
De acordo com o artigo, a deficiência do nutriente está relacionada a doenças hepáticas, pode comprometer a função cognitiva dos descendentes e causar possíveis distúrbios neurológicos.
Planejamento é essencial
Derbyshire alerta que quem adota uma alimentação vegana precisa compensar a possível deficiência do nutriente - assim como de ferro, vitamina B12, ômega-3 e cálcio.
"Se você não gosta desses alimentos, pode precisar tomar suplementos."
Mas a Associação Dietética Britânica (BDA, na sigla em inglês) afirma que, com planejamento, é possível obter quantidade suficiente de colina a partir de uma dieta vegana.
"É perfeitamente possível atender a esses requisitos com uma dieta vegana ou à base de plantas", declarou Bahee Van de Bor, porta-voz da BDA.
"Mas você precisa se planejar. Os alimentos podem ser veganos, mas não fornecer os nutrientes necessários."
Os veganos devem prestar atenção, portanto, no que consomem e assegurar uma dieta variada.
"É provável que uma dieta vegetariana ou vegana saudável e variada forneça um pouco de colina", diz o porta-voz.
Também é importante garantir que essa dieta seja bem equilibrada para garantir a ingestão de nutrientes suficientes como ferro, cálcio, zinco e vitamina B12.
"Dito isto, sabemos que pode haver muitos benefícios para a saúde ao seguir uma dieta baseada mais em plantas, embora isso não signifique necessariamente que os produtos de origem animal devam ser completamente excluídos", acrescentou.
Segundo Derbyshire, pesquisas indicam que mulheres grávidas e lactantes, em particular, precisam garantir ingestão suficiente de colina em suas dietas, uma vez que o nutriente é extremamente importante para o desenvolvimento cerebral do feto.
"Quero, em primeiro lugar, aumentar a conscientização. Mas também acho que se as pessoas estão adotando uma alimentação à base de verduras e legumes, principalmente mulheres em idade fértil, devem buscar suplementos", afirmou.
A Primeira Lei de Mendel ou Lei da Segregação dos Fatores determina que cada característica é condicionada por dois fatores que se separam na formação dos gametas.
A segregação é consequência da localização dos genes nos cromossomos e do comportamento desses durante a formação dos gametas, através do processo de meiose.
O monge Gregor Mendel realizou seus estudos com objetivo de compreender como as diferentes características eram transmitidas de uma geração para outra.
Experimentos com Ervilhas
Gregor Mendel conduziu seus experimentos utilizando ervilhas pelos seguintes motivos:
Planta de fácil cultivo e desenvolvimento em curto período;
Produção de muitas sementes;
Rápido ciclo reprodutivo;
Facilidade de controlar a fecundação das plantas;
Capacidade de realizar autofecundação.
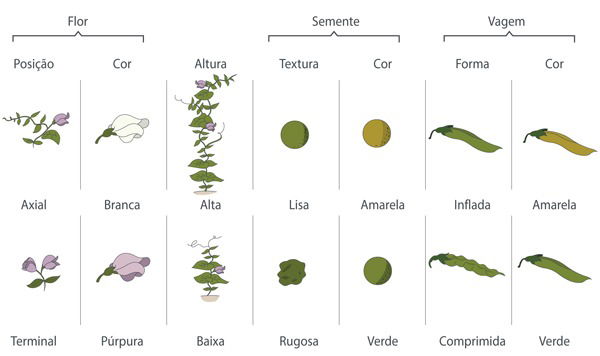
Os seus experimentos analisaram sete características das ervilhas: cor da flor, posição da flor no caule, cor da semente, textura da semente, forma da vagem, cor da vagem e altura da planta.
Ervilhas e as características estudadas por Gregor Mendel em seus experimentos genéticos
Ao observar a cor das sementes, Mendel percebeu que a linhagem de sementes amarelas sempre produziam 100% dos seus descendentes com sementes amarelas. E o mesmo acontecia com as sementes verdes.
As linhagens não apresentavam variações, constituindo linhagens puras. Ou seja, as linhagens puras mantinham suas características ao longo das gerações.
Os achados de Gregor Mendel são considerados o marco inicial para os estudos genéticos. A sua contribuição para a área foi imensa, o que levou a ser considerado o "pai da Genética".
Cruzamentos
Como estava interessado em saber como as características eram passadas de uma geração para outra, Mendel realizou outro tipo experimento.
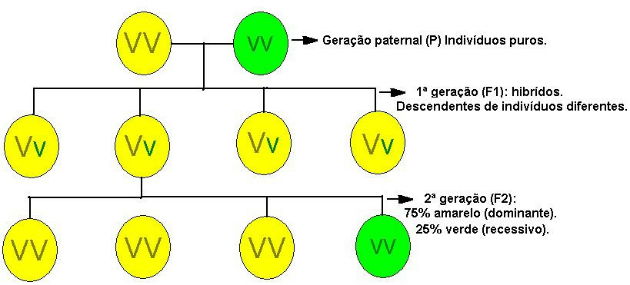
Dessa vez, realizou o cruzamento entre linhagens puras de sementes amarelas e sementes verdes, o que constituiu a Geração Parental.
Como resultado desse cruzamento, 100% das sementes eram amarelas - Geração F1.
Mendel concluiu que a semente amarela apresentou dominância sobre a semente verde. Surgia, assim, o conceito de genes dominantes e recessivos na genética.
Como todas as sementes geradas eram amarelas (Geração F1), Mendel realizou a autofecundação entre elas.
Os resultados surpreenderam Mendel, na nova linhagem (Geração F2) surgiram novamente as sementes verdes, na proporção 3:1 (amarelas:verdes). Ou seja, foi observado que a cada quatro plantas, três apresentavam a característica dominante e uma a característica recessiva.
Cruzamentos da Primeira Lei de Mendel
Mendel concluiu que a cor das sementes era determinada por dois fatores: um fator para gerar sementes amarelas, que é dominante, e outro fator para gerar sementes verdes, recessivo.
Assim, a 1ª Lei de Mendel pode ser enunciada como a seguir:
“Todas as características de um indivíduo são determinadas por genes que separam-se, durante a formação dos gametas, sendo que, assim, pai e mãe transmitem apenas um gene para seus descendentes”.
Primeira e Segunda Lei de Mendel
A Primeira Lei de Mendel diz que cada característica é condicionada por dois fatores que se separam na formação dos gametas.
Nesse caso, Mendel estudou apenas a transmissão de uma única característica. Por exemplo, cruzou sementes amarelas com sementes verdes.
A Segunda Lei de Mendel baseia-se na transmissão combinada de duas ou mais características. Por exemplo, ele realizou cruzamentos de sementes verdes e rugosas com sementes amarelas e lisas.
Em conjunto, as Leis de Mendel explicam como as características hereditárias são transmitidas de uma geração à outra.
Por meio dos estudos de cruzamento de plantas com características diferentes foi possível comprovar que as mesmas mantém sua integridade ao longo das gerações.
Exercício Resolvido
1. (FUC-MT) Cruzando-se ervilhas verdes vv com ervilhas amarelas Vv, os descendentes serão: a) 100% vv, verdes; b) 100% VV, amarelas; c) 50% Vv, amarelas; 50% vv, verdes; d) 25% Vv, amarelas; 50% vv, verdes; 25% VV, amarelas; e) 25% vv, verdes; 50% Vv, amarelas; 25% VV, verdes.
Resolução
Para resolver a questão deve-se realizar o cruzamento entre as ervilhas verdes recessivas (vv) e ervilhas amarelas heterozigóticas dominantes (Vv):
Vv x vv → os genótipos originados são: Vv Vv vv vv Logo, temos 50% de Vv (ervilhas amarelas) e 50% vv (ervilhas verdes).
Resposta: Letra c) 50% Vv, amarelas; 50% vv, verdes.
1. (Unifor-CE) Um estudante, ao iniciar o curso de Genética, anotou o seguinte:
I. Cada caráter hereditário é determinado por um par de fatores e, como estes se separam na formação dos gametas, cada gameta recebe apenas um fator do par. II. Cada par de alelos presentes nas células diploides separa-se na meiose, de modo que cada célula haploide só recebe um alelo do par. III. Antes da divisão celular se iniciar, cada molécula de DNA se duplica e, na mitose, as duas moléculas resultantes se separam, indo para células diferentes. A primeira lei de Mendel está expressa em:
a) I, somente. b) II, somente. c) I e II, somente. d) II e III, somente. e) I, II e III.
Bióloga, apaixonada por ensino. Fascinada por ciências forenses, meio ambiente ,leis, design, psicologia e medicina legal. Cada dia aprendendo um pouco e compartilhando com você.
Obrigada por estar aqui.













 Imprimir
Imprimir










